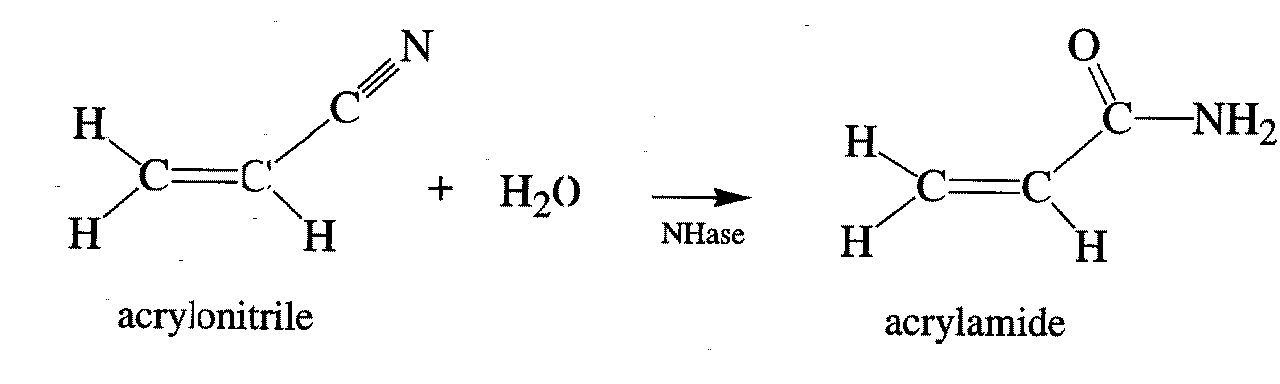 Fig.1. Reaction catalyzed by nitrile hydratase.
Fig.1. Reaction catalyzed by nitrile hydratase. Extensible Systematic Force Field Study of Photoactive Protein Nitrile Hydratase

 Contents:
Contents:
Keywords: Nitrile Hydratase, Molecular Dynamics, ESFF, Biotechnology
Nitrile Hydratase (Nhase, EC. 4.2.1.84) is an enzyme used in biotechnological industrial production of acrylamide. Recently its structure has been elucidated [1]. The active site containing non-heme iron is buried in the protein core just at the interface of two domains. Hydrogen bonds between Arg56 (subunit B) and Cys114 sulfenic acid (subunit A) are important to maintain the activity of the protein. In order to propose a plausible mechanism of the catalytic activity possible structural changes of the active site should be studied [2]. We have recently found that the iron may undergo "inverted doming" after the nitric oxide ligand photodissociation [3]. In this work results of the ESFF modeling of two domain Nhase are presented. In particular, an interaction of 1,4-dioxane with the entry channel is analyzed. The molecular dynamics study using the DISCOVER module of InsightII (Accelrys) may provide data for calculations of substrat/product reaction paths in this important enzyme. Preliminary results of 1ns NHase dynamics obtained using MOIL forcefield [4] are also presented.
Introduction.
Biotechnology bings a great promise for a new ways of doing chemistry. In particular industrial production of certain chemicals may be achived without charmful and expensive processes. A good example of the advantages of using microorganisms in chemistry is production of acrylamide. Japaneese companies use bacteria Rhodococcus Sp. for routine production of kilotons of this commodity. The reaction (see Fig. 1) is catalysed by the protein nitrile hydratase (NHase).
Fig.1. Reaction catalyzed by nitrile hydratase.  Fig.2 NHase structure.
Fig.2 NHase structure.
Methods
The initial structure of NHase was adopted from PDB - file 2AHJ.
The ESFF forcefield (see for example [ 7 and references therein] )as implemented in InsightII v. 97.0 package [6] was chosen for its ease in parametization. It is a diagonal forcefield, with Morse bond potentials and based on density functional theory calculations of atomic electronegativity and hardness. The charges are determined in complex way, by minimizing the electrostatic energy under the constraints that the sum of charges is equal to the net charge of the molecule. In our model the system have a charge of -23 since no counterions were used on this stage of the study. The ESFF uses the 6-9 potential for the van der Waals interactions. In this forcefield, van der Waals parameters are affected by the charge of the atom. An important part of modeling is correctly setting up all the bond orders in the system under study.
All crystallographic water molecules were retained in the model (>200). Hydrogen atoms were added using the "Biopolymer" module of the InsightII package. The Builder module was used to substitute Cys112 and Cys114 for CSD and CEA. One should note, that standard ESFF s4l and s3d sulfur types not necessary describe correctly the geometry of the active site.
For molecular dynamics (MD) simulations, a small cutoff of 9.5 Ang. was used. The structure was subject to 200 steps of minimization before the 15 ps heating stage and 50 ps quillibration MD runs. A dielectric constant of 1 was used through all computations. All bond lengths were kept constant using RATTLE algorithm. The simulation timestep was 1 fs. The initial temperature was 10 K. During the dynamics simulations it was kept constant at 298 K by a velocity scaling. The sampling data were collected through 5 ps, each 0.1 ps, and analyzed using InsightII 97.0. Calculations were performed on the SGI R5000 workstation.
The MOIL model of Nitrile hydratase was construted in the following way. The crystal structure of an inactive form of nitrile hydratase isolated from Rhodococcus sp.N-771 (2ahj.pdb, resolution 1.70A) [1] was obtained from the Protein Data Bank. All minimizations, heatings, equilibrations, dynamics runs and the analysis were performed using the package MOIL (ver. 7.1) [4] and Moil-View (ver. 9.0) [4a] software running on a Silicon Graphic Indy R5000 computer. The active site of Nhase contains four residues: Cys109, CSD112 (Cys-SO2H), Ser113 and CEA114 (Cys-SOH). The central atom, Fe(III) is coordinated by sulfur atoms of Cys109, CSD112 and CEA114, and two other coordinating atoms are amide nitrogens of Ser113 and CEA114. The sixth coordinating position is occupated by nitrogen atom of nitric oxide (NO). In order to perform molecular dynamics simmulation we prepared a new, non-standard residue called BFE (BigFe), representing the Fe-type active site of Nhase. This residue contained: Val108, Cys109, Ser110, Leu111, CSD112, Ser113, CEA114, Thr115, and the Fe(III) atom and was built by adding 68 new atom types. We described all properties of those atoms, eg. masses, charges and van der Waals parameters, and in order to construct from them the BFE residue, we have defined 71 bonds, 109 bond angles, 43 torsions and 31 improper torsions. Masses and van der Waals parameters of atoms were standardi (i.e. as used in the MOIL forcefield), charges of atoms of Val108, Ser110, Leu111 and Thr115 were standard too, but charges of atoms belonging to CSD112, Ser113 and CEA114 were the same as we the Mulliken charges caculated with the DFT method. These calculations we presented elsewere [3].
MOIL MD protocol.
Initially hydrogen atoms were addedto the bare pdb the Nhase molecule. Crystallographics waters were retained. A system was subject to 3000 steps of minimization by the Powell method [4]. Subsequently the heating (10-300K) step was done through 20ps and then equilibration at 300K through 30 ps. Data were collected each 100 steps through 950 ps period of simulation of the dynamics. We kept the constant temperature by the velocity scaling. Cutoff distances for electrostatic and Van der Waals interactions were of 12 and 7 Ang., respectively. To reduce a time step to 1 fs all bonds in the protein and all bonds and angles in water molecules were kept fixed by the SHAKE protocol.
Results and Discussion
The ESFF forcefield
It is known that the RMS value is a proper tool to estimate the stability of protein structure during a simulation. Thus in the first step of analysis the dynamical behaviour of the nitrile hydratase model we checked the RMS value vs. time (Fig.3). As expeceted, in such rather short simulation a full equillibration is not achieved, but the RMS tend to be rather stable.
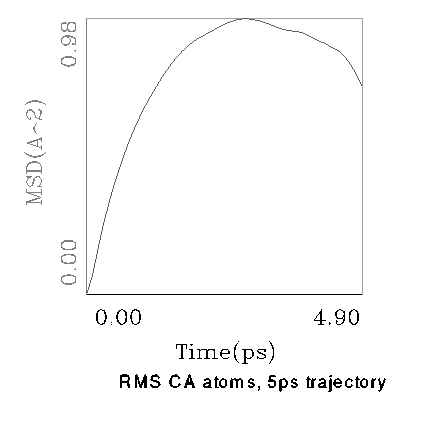 Fig.3. The RMS value for CA atoms calculated during 5ps
period of dynamics.
Fig.3. The RMS value for CA atoms calculated during 5ps
period of dynamics. Water is involved in postulated mechanism of the catalysis. We were looking for water molecules having locations close to the iron site. Such water might occupate the 6'th coordinate position after the dissociation of the NO molecule. Since in the radius of about 4 to 6 Ang. from the iron atom we did not find any water molecule (data not shown), perhaps DIO molecule effectively blocks an access to the active site for larger molecules. One should note that our statistics is very limited, so in longer trajectories some water presence in close proximity of Fe may occur, especially in the active forms of NHase.
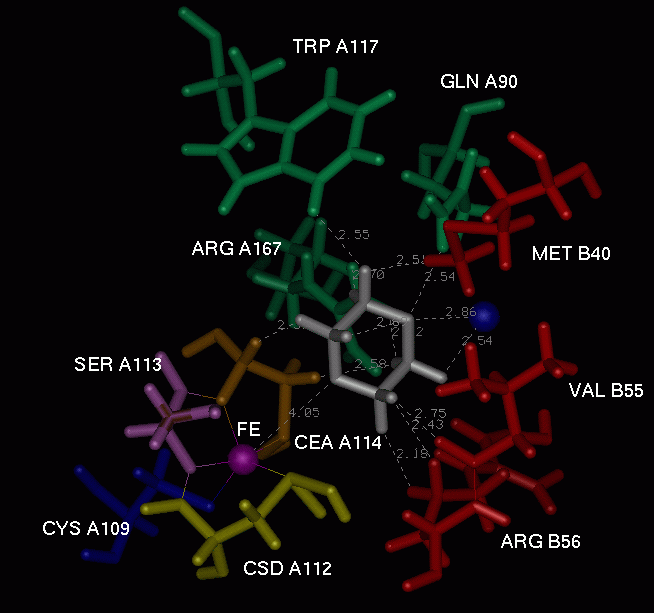 Fig.4. The environment of the DIO molecule. Distances to the nearest
neighbours are also indicated.
Fig.4. The environment of the DIO molecule. Distances to the nearest
neighbours are also indicated.
Table 1. The distances for the nearest neighbours of the DIO molecule. Min, max and average distances between two atoms, wchich could be involved in the hydrogen bonds are listed. Distances for PDB structure are also indicated. In this case the hydrogen atoms were added using the BIOPOLYMER module of the INSIGHT package ver. 95.0 [6].
|
Ligand atom - NHase |
Dist. PDB [1] |
Min. dist. |
Max. dist |
Aver. |
|
DIO:H21 - GlnA90:NE2 |
7.68 |
2.43 |
5.86 |
4.58 |
|
DIO:O1 - GlnA90:HE21 |
6.88 |
2.32 |
7.15 |
5.67 |
|
DIO:O1 - GlnA90:HE22 |
6.86 |
2.24 |
5.75 |
4.24 |
|
DIO:O1 - GlnA90:HG1 |
5.49 |
5.26 |
9.03 |
7.08 |
|
DIO:O1 - GlnA90:HG2 |
6.48 |
3.78 |
8.75 |
2.15 |
|
DIO:O1' - SerA113:HG |
5.57 |
3.25 |
5.87 |
4.20 |
|
DIO:O1' - CEA114:HA |
5.43 |
2.84 |
5.15 |
3.87 |
|
DIO:O1 - CEA114:HB1 |
8.25 |
3.08 |
6.77 |
5.01 |
|
DIO:O1 - TrpA117:HZ2 |
3.66 |
2.51 |
5.34 |
4.23 |
|
DIO:H11 - ArgA167:NH2 |
6.89 |
2.89 |
8.98 |
6.17 |
|
DIO:H12 - ArgA167:NH2 |
7.45 |
3.91 |
10.09 |
7.44 |
|
DIO:O1 - ArgA167:HH22 |
7.38 |
3.56 |
8.18 |
6.26 |
|
DIO:O1 - MetB40:HE2 |
4.45 |
2.62 |
5.07 |
4.01 |
|
DIO:O1 - MetB40:HE3 |
4.72 |
2.54 |
6.62 |
4.69 |
|
DIO:H1'2 - ArgB55:N |
9.47 |
5.36 |
7.93 |
6.29 |
|
DIO:O1 - ArgB55:HB |
5.48 |
3.08 |
6.35 |
4.42 |
|
DIO:O1' - ArgB56:HG2 |
4.03 |
3.69 |
6.12 |
5.03 |
|
DIO:H1'1 - ArgB56:NE |
5.94 |
3.78 |
8.57 |
6.34 |
|
DIO:H12 - ArgB56:NE |
2.59 |
2.60 |
8.61 |
6.12 |
|
DIO:H1'2 - ArgB56:NE |
5.25 |
3.14 |
7.52 |
4.97 |
|
DIO:H12 - ArgB56:NH2 |
6.36 |
3.67 |
9.16 |
7.18 |
|
DIO:O1 - ArgB56:HE |
5.96 |
3.68 |
7.63 |
6.15 |
|
DIO:H12 - NO:O |
3.83 |
2.31 |
6.15 |
4.59 |
To get a better insight into dynamical behaviour of the DIO molecule and to estimate possible positions for docking of this moiety we monitored distances between the DIO, FE iron and one of mentioned residues eg. ValB55. As one can see (Fig. 5) the amplitude of motions of DIO in respect to the position of FE(III) ion is about 1 Ang. (Fig. 5a and 5b) so we can say that position of DIO molecule is rather stable. As we expected, the amplitude of relative motions DIO vs. ValB55 are not so small, this is probably caused by the overlap of two kinds of motions: motions of the DIO molecule and motions of the side chain of ValB55 residue.
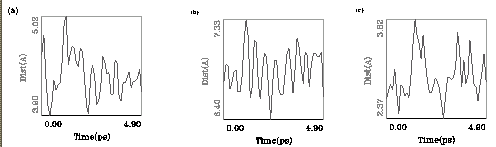 Fig.5. Distances in the environment of the DIO molecule (ESFF 5 ps data). Showed distances: (a) FE - O1' DIO
(b) FE - O1 DIO and (c) H1' DIO - O ValB55.
Fig.5. Distances in the environment of the DIO molecule (ESFF 5 ps data). Showed distances: (a) FE - O1' DIO
(b) FE - O1 DIO and (c) H1' DIO - O ValB55.The MOIL forcefield
For the long 1 ns trajectory obtained with the MOIL forcefield plots of some RMS values are also presented. The RMS calculated for CA atoms (all protein) achieved the maximum value of about 6.5 Ang. (Fig. 6). Such a high value of the RMS could suggest that the structure of NHase is not well maintained in this forcefield. On the other hand long trajectory data may reveal intersiting structural transformations related to the activity of the enzyme. In order to check which regions of the NHase were undergoing the largest structural rearrangements, i we calculated the RMS for subunit A only (Fig. 6 and Fig. 7),for subunit B only (Fig. 6 and Fig. 7), for domain A of subunit B and for domain B of subunit B (Fig. 7).
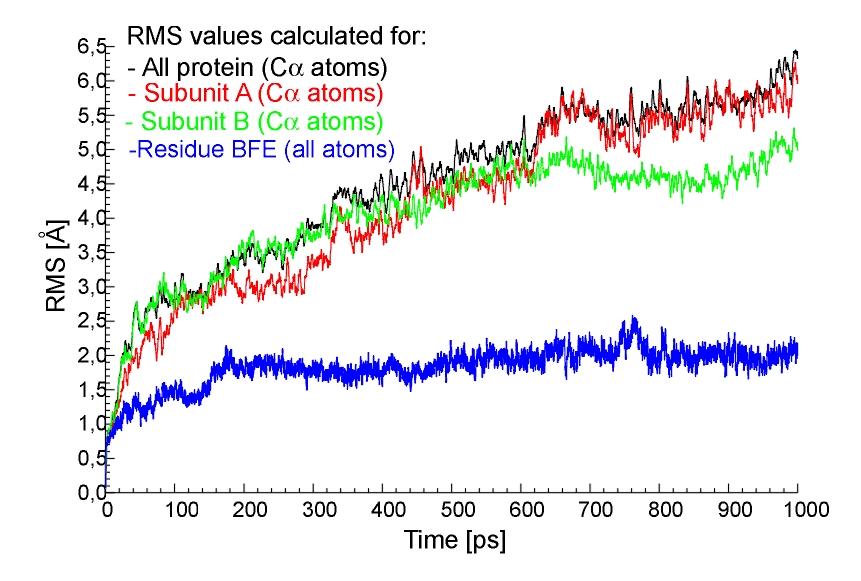 Fig.6. The RMS values for CA atoms from all protein (black line),
subunit A only (red line), subunit B only (green line) and for all atoms formnig the BFE residue (blue line), 1ns trajectory, MOIL.
Fig.6. The RMS values for CA atoms from all protein (black line),
subunit A only (red line), subunit B only (green line) and for all atoms formnig the BFE residue (blue line), 1ns trajectory, MOIL. 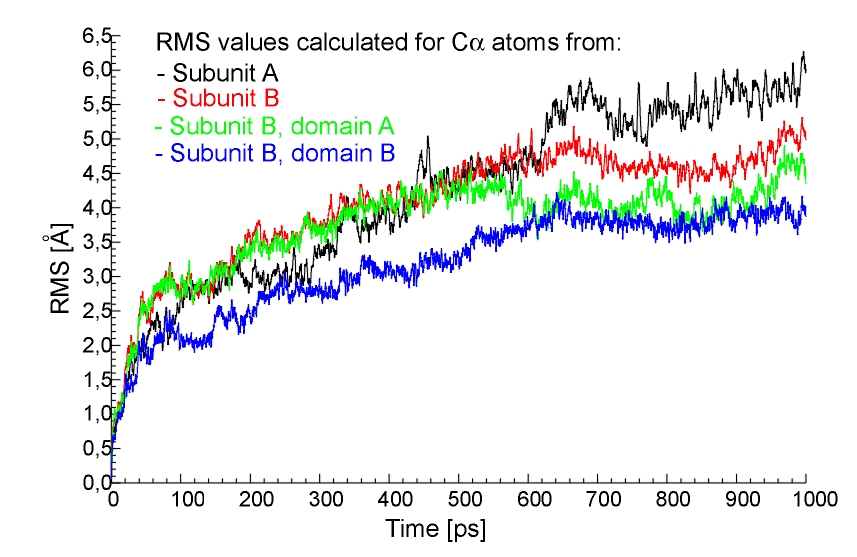 Fig.7. The RMS values for CA atoms from all subunit A (black line),
all subunit B (red line), domain A of subunit B (green line) and domain B of subunit B (blue line), 1ns trajectory, MOIL.
Fig.7. The RMS values for CA atoms from all subunit A (black line),
all subunit B (red line), domain A of subunit B (green line) and domain B of subunit B (blue line), 1ns trajectory, MOIL. In this case the maximum value of the RMS is about 2.5 Ang. This result indicates the stability of this model of active site; RMS is rather small despite it includes possiblity for a relatively large side chains motions. Thus this is the supporting evidence that our parameters, including partial charges calculated elsevere [3], work.
It is known that three oxygen atoms in the active site (OG Ser113, OD1 CSD112 and OD CEA114) have unusual triangular arrangement and forms so-called "oxygen claw setting". These oxygen atoms were postulated to play an important role in the stabilization of binding of NO molecule to the NHase [1]. Insight into that interactions could be provided by the monitoring distances between the mentioned oxygen atoms. As shown in Fig. 8 this distances are higher than those observed in the X-ray. Since doing parametrization of the BFE we were trying to keep as many original MOIL parameters as possible, and we did not introduced any additionas constrains to maintain the experimenatlly observed oxygen claw setting, this observation indicated that subtle crystal packing may be required to keep affectively these 3 oxygens in such a close proximity. It would be very useful to know an Xray structure of the active form of NHase with the properly oxidized Cys112 and Cys114 residues. The low resolution structure reported by Huang et al. ( pdb code 1AHJ ) has aparently a wrong interpretation of the active site electron density. On the other hand this observation make us to think that our model of active site is not ideal. Studies on the parametrization of BFE/MOIL "residue" are necessary.
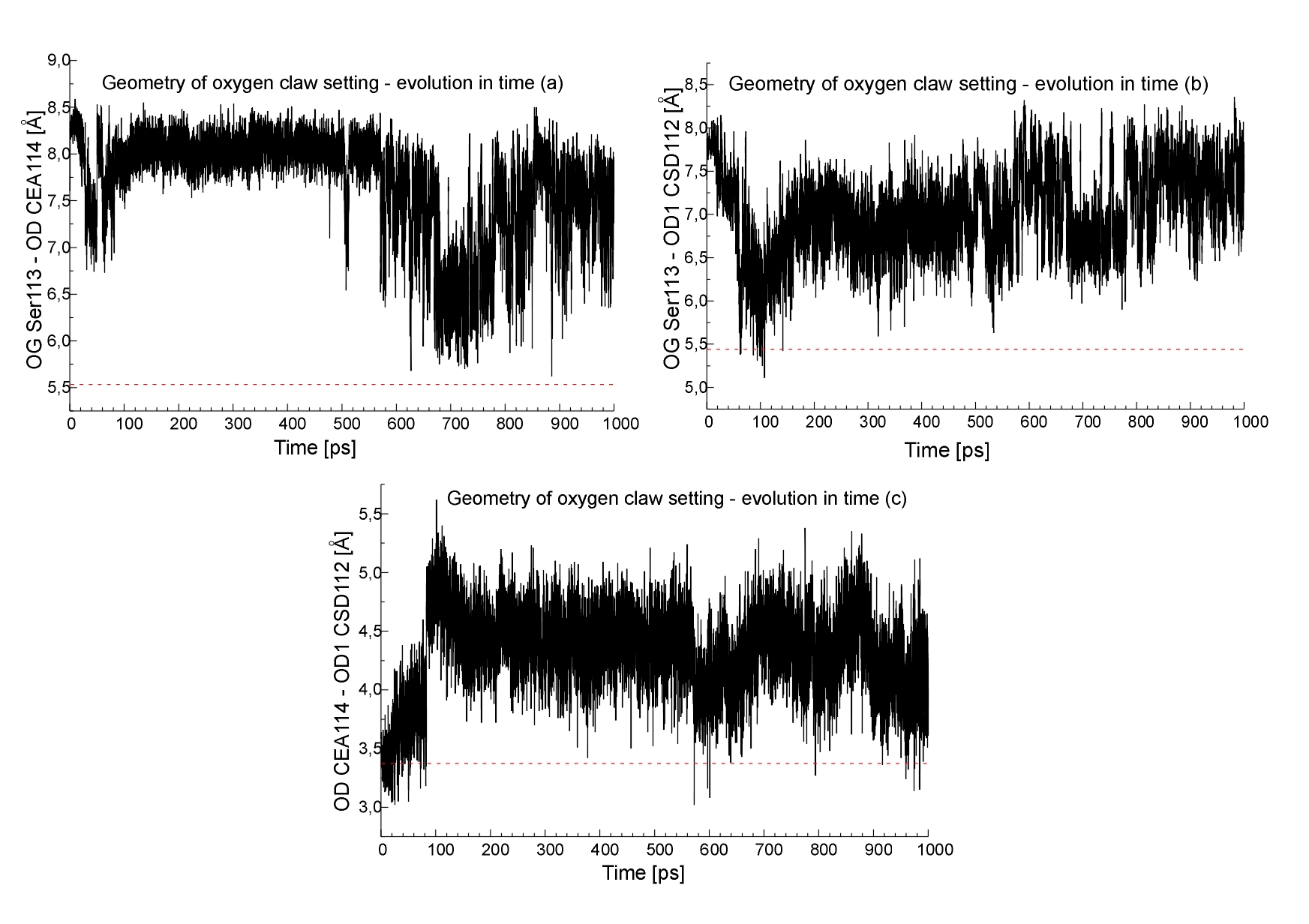 Fig.8. The geometry of the oxygen atom forming so-called
"oxygen claw setting",(a) OG Ser113:OD CEA114 (b) OG Ser113:OD1 CSD112 (c) OD CEA114:OD1 CSD112 distances, geometry for the PDB structure
is also indicated (red line).
Fig.8. The geometry of the oxygen atom forming so-called
"oxygen claw setting",(a) OG Ser113:OD CEA114 (b) OG Ser113:OD1 CSD112 (c) OD CEA114:OD1 CSD112 distances, geometry for the PDB structure
is also indicated (red line).
Conclusions
The ESFF forcefield gives rather reasonable quality results for the geometry of the nitrile hydratase and therefore might be used as an alternative approach to the modeling studies of this enzyme. However the structure of the iron active center is not correctly maintained, further studies on parametrization of this region are required. The problem is related to the proper description of non-standard sulfenic and sulfinic acid residues. The dioxane molecule is kept in its position by a network of steric interactions. We were not able to indicate any particular hydrogen bonds that might be related to dioxane binding in the entry chanel of NHase. In our ESFF model, the NO molecule could diffuse out of the Fe centre on the 50 ps timesale (300 K), despite the presence of the dioxane "flap". Our data indicate that the environment of the dioxane molecule indentified here (Fig. 4) could play a role in the catalytic activity of the enzyme and should be studied in grater detail. Mutations in this region, especially of charged residues such as Arg56 might affect NHase performance.
The geometry of the NHase and particularity the structure and interacions in the neighborhood of the active site were studied using the MOIL forcefield. For the first time the active site model of NHase was constructed in this forcefield. Our parameters of the atoms included in the BFE residue reflected stability during the 1ns trajectory (including the equillbration and heating period of our simulation procedure). This reasonable stability is indicated in Fig. 7 and Fig.9.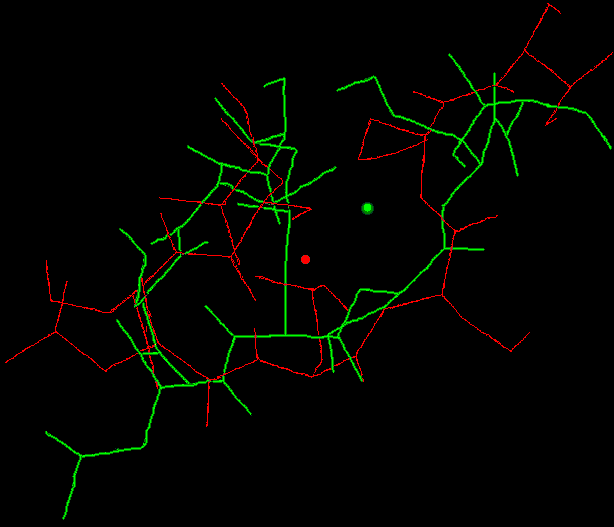 Fig.9. The overlap of non-standard BFE residue: the starting
geometry (red) and the structure after 750ps (green).
Fig.9. The overlap of non-standard BFE residue: the starting
geometry (red) and the structure after 750ps (green). 


 Fig.10. The overlap of CA atoms from all protein. The starting geometry is indicated by red colour, the geometries after 250ps (A), 500ps (B)
750ps (C) and 1ns (D) are indicated by green.
Fig.10. The overlap of CA atoms from all protein. The starting geometry is indicated by red colour, the geometries after 250ps (A), 500ps (B)
750ps (C) and 1ns (D) are indicated by green.
References
1. S. Nagashima et al., Nature Struct. Biol. 1998, 5, 347.
2. P.K. Mascharak, Coord. Chem. Rev. 2002, 225, 201.
3. W. Nowak et al., Int. J. Quantum Chem. 2002, 90, 1174.
4. R. Elber et al., Comput. Phys. Comm., 1994, 91, 159.
4a. C. Simmerling, R.Elber et al., in "Modeling of Biomolecular`Structure and Mechanism", A.Pullman et al. eds., 241-261, Kluwer, 1995.
5. A. Boone et al., Inorg. Chem. 2001, 40, 1837.
6. InsightII, Accellrys USA, 1997.
7. W. Nowak et al., Acta Bioch. Pol., 2001, 48, 903.